在新型含能材料的性能预估中,爆轰性能是重中之重。只有仿真的数据足够引人重视,研究人员才会投入人力物力财力去进行实际测试。而爆轰性能是在密度、生成焓、分子组成等基础上,通过经验公式或爆轰状态方程分析计算得到的。在之前的文章里,我已经介绍过如何使用ChemBioOffice 2014、Gaussian 03w、Multiwfn等软件,来预估中性分子及离子盐的晶体密度。现在我将通过本文,初步介绍一下如何计算离子盐的晶格焓与生成焓。
1、晶格能与晶格焓
含能离子的生成焓可根据方程(1)得出:
ΔHf0 (离子盐,298K) = ΔHf0 (阳离子,298K) + ΔHf0 (阴离子,298K) - ΔHL (1)
其中ΔHL 为离子盐的晶格焓。
ΔHL 根据方程(2)[1]求出:
ΔHL = UPOT + [p (nM/2-2) + q (nX/2-2)] R T (2)
其中UPOT为晶格能;p、q分别为阳离子与阴离子的电荷数;nM和nX由阳离子和阴离子的结构性质决定,单原子离子取3,线性多原子离子取5,非线性多原子离子取6;R为理想气体常数;T为绝对温度。
UPOT根据方程(3)[1]求出:
UPOT = γ(ρm/Mm)1/3 + δ (3)
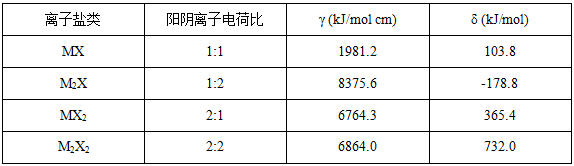
γ和δ是根据阳阴离子的比值而变化的常数,具体取值见表1-1。
表1-1 方程(3)中的常数取值
![Snap2.jpg]()
以上计算简易,精度尚可,不需要使用量子化学计算软件,仅使用密度和分子量在excel上便能快速得出结果。
2、生成焓
生成焓的计算较为复杂,需要先求出阳阴离子的生成焓。而离子盐阳阴离子的生成焓则要引入质子亲合势(proton affinity)这一参数。质子亲合势的定义是标准状态下分子接受质子的能力。需要经Gaussian软件求得阳阴离子的系统总电子能量(Eele)和零点振动能(ZPE),计算方程[2]为:
PA = - ΔH
= - ΔEele - ΔZPE + 5/2RT
= E0(A) - E0(AH+) + 5/2RT (4)
其中E0为经过零点能校正后的能量,单位为Hartree可通过(5)换算为kJ或kcal:
1 Hartree = 27.2 ev = 627.5 kcal = 2625.5 kJ (5)
通过软件获得质子亲合势后,可根据方程(6)、(7)求得阳阴离子生成焓:
ΔHf0 (阳离子,298K) = ΔHf0 (阳离子对应分子,298K,g) - PA(阳离子) (6)
ΔHf0 (阴离子,298K) = ΔHf0 (阴离子对应分子,298K,g) + PA(阴离子) (7)
要注意的是质子亲合势为负表示结合质子,运算时根据离子是否获得质子调整正负。
3、计算实例
由于仅一个离子就需要运行两次软件分别获得E0和分子气相生成焓,加上后续使用excel进行数学计算。步骤多且繁杂,因此本文只使用硝酸铵一个实例来阐明计算过程。
1、根据硝酸铵分子式为NH4NO3属于MX型离子盐,查阅表1-1选取常数γ为1981.2,常数δ为103.8。代入方程(3)求得:
UPOT = 654.702 kJ/mol
2、根据NH4+和NO3-的离子形态,选取p、q均为6,将结果代入方程(2)求得:
ΔHL = 659.658 kJ/mol
3、运行ChemBioOffice 2014中对接的MOPAC,使用PM7半经验分子轨道方法分别计算NH3和HNO3的气相生成焓,结果为:
ΔHf0 (NH3,298K,g) = -17.8 kJ/mol
ΔHf0 (HNO3,298K,g) = -143.4 kJ/mol
4、运行Gaussian 03w选择frequency任务类型,使用密度泛函B3LYP,设置为6-311++G(2d,2p)基组。分别计算NH3和NH4+,运行结束后打开输出文件,找到Sum of electronic and zero-point Energies一栏,即得到方程(4)中的E0(A)和E0(AH+)。结果为:
E0(NH3) = -56.55 Hartree
E0(NH4+) = -56.87 Hartree
同样运行后得:
E0(HNO3) = -280.91 Hartree
E0(NO3-) = -280.44 Hartree
将以上结果代入方程(4)得:
PA(NH4+) = 851.14 kJ/mol
PA(NO3-) = 1235.98 kJ/mol
5、将步骤3结果和质子亲合势代入方程(6)、(7)求出阳阴离子的气相生成焓:
ΔHf0 (NH4+,298K) = -868.94 kJ/mol
ΔHf0 (NO3-,298K) = 1092.58 kJ/mol
6、最后根据方程(1)得出硝酸铵的生成焓:
ΔHf0 (硝酸铵,298K) = -436.01 kJ/mol
硝酸铵的实测生成焓为-365.6 kJ/mol,计算值较实际值相差70.41 kJ/mol。
4、结束语
本文重点是论述整个计算过程,实际上还可以采用先优化构型再计算和选择精度更高的基组等方法来大幅提高仿真精度。还有就是本文的方法是仓促中从搜集的资料里摸索出来的,难免会出现一些错误,在真正搞量子化学计算的人眼里看来也是班门弄斧。我的希望就是感兴趣的朋友能在我的基础上进一步的将含能材料仿真体系建立起来,甚至能取得成果得到专业人士的认可,那么我的些许付出就是很值得的了。
虽然大厦的基石已经铺好,但是行百者半九十,任重而道远,与诸君共勉
[1] H D Jenkins,D Tudela,L Glasser. Lattice Potential Energy Estimation for Complex Ionic Salts from Density Measurements.[J].Inorg Chem,2002,41(9):2364-2367.
[2] 鲁礼林. 密度泛函理论中经验参数的优化筛选[博士学位论文D]. 武汉:武汉大学,2012.
更详细内容见附件
 含能离子晶格焓与生成焓的计算.docx
20.10KB
DOCX
155次下载
含能离子晶格焓与生成焓的计算.docx
20.10KB
DOCX
155次下载




200字以内,仅用于支线交流,主线讨论请采用回复功能。