
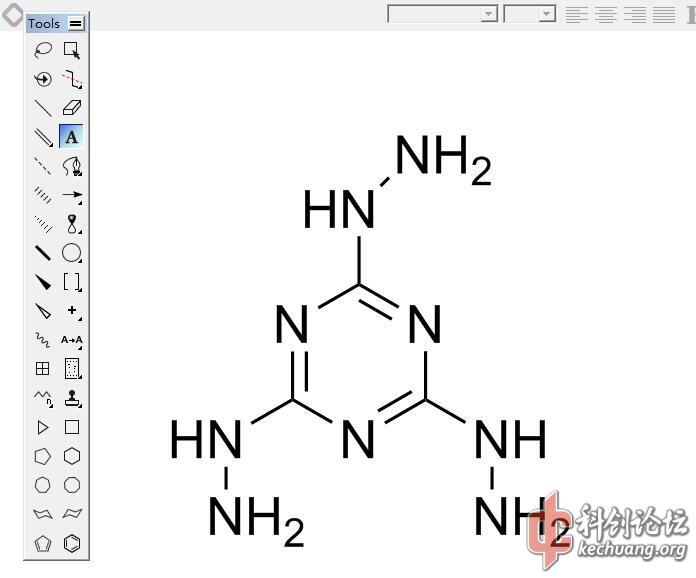






过程很详尽,对于这种基组和算法,结果偏差已经算是在合理范围了。。
至于化学键消失的问题,其实不用怎么在意,键连是通过计算机判断键长来确定的,所以可能会因此判断为无化学键
至于化学键消失的问题,其实不用怎么在意,键连是通过计算机判断键长来确定的,所以可能会因此判断为无化学键
[修改于 7年9个月前 - 2016/08/31 15:15:39]
引用 718281828kc:原来是这个原因,我一直以为是软件对接出错了。。。下一步就是看看把误差再缩小一点达到能指导应用的地步。
过程很详尽,对于这种基组和算法,结果偏差已经算是在合理范围了。。
至于化学键消失的问题,其实不用怎么在意,键连是通过计算机判断键长来确定的,所以可能会因此判断为无化学键
我也测试了一下。分别使用B3LYP/6-31G**和M06-2X/def2TZVP进行几何优化并计算5-氨基四唑的密度。结果如下:
B3LYP/6-31G**
Volume: 630.00144 Bohr^3 ( 93.35660 Angstrom^3)
Estimated density according to mass and volume: 1.5131 g/cm^3
Minimal value: -36.60195 kcal/mol Maximal value: 54.97392 kcal/mol
Overall surface area: 387.10628 Bohr^2 ( 108.40082 Angstrom^2)
Positive surface area: 173.08492 Bohr^2 ( 48.46872 Angstrom^2)
Negative surface area: 214.02137 Bohr^2 ( 59.93210 Angstrom^2)
Overall average value: 0.00029148 a.u. ( 0.18291 kcal/mol)
Positive average value: 0.03376935 a.u. ( 21.19061 kcal/mol)
Negative average value: -0.02678298 a.u. ( -16.80659 kcal/mol)
Overall variance (sigma^2_tot): 0.00075443 a.u.^2 ( 297.06915 (kcal/mol)^2)
Positive variance: 0.00052564 a.u.^2 ( 206.98141 (kcal/mol)^2)
Negative variance: 0.00022878 a.u.^2 ( 90.08774 (kcal/mol)^2)
Balance of charges (miu): 0.21129146
Product of sigma^2_tot and miu: 0.00015940 a.u.^2 ( 62.76817 (kcal/mol)^2)
Internal charge separation (Pi): 0.02993833 a.u. ( 18.78660 kcal/mol)
0.9138*1.5131+0.0028*62.76817+0.0443=1.603
M06-2X/TZVP
Volume: 639.89024 Bohr^3 ( 94.82197 Angstrom^3)
Estimated density according to mass and volume: 1.4897 g/cm^3
Minimal value: -36.45772 kcal/mol Maximal value: 58.45159 kcal/mol
Overall surface area: 392.17047 Bohr^2 ( 109.81893 Angstrom^2)
Positive surface area: 184.59652 Bohr^2 ( 51.69230 Angstrom^2)
Negative surface area: 207.57395 Bohr^2 ( 58.12663 Angstrom^2)
Overall average value: 0.00158584 a.u. ( 0.99513 kcal/mol)
Positive average value: 0.03332935 a.u. ( 20.91450 kcal/mol)
Negative average value: -0.02664382 a.u. ( -16.71926 kcal/mol)
Overall variance (sigma^2_tot): 0.00078132 a.u.^2 ( 307.66095 (kcal/mol)^2)
Positive variance: 0.00056427 a.u.^2 ( 222.19344 (kcal/mol)^2)
Negative variance: 0.00021705 a.u.^2 ( 85.46751 (kcal/mol)^2)
Balance of charges (miu): 0.20062615
Product of sigma^2_tot and miu: 0.00015675 a.u.^2 ( 61.72483 (kcal/mol)^2)
Internal charge separation (Pi): 0.02990414 a.u. ( 18.76514 kcal/mol)
0.9138*1.4897+0.0028*61.72483+0.0443=1.578
两个结果与实验值的差别不是很大。我在想是不是楼主在计算的时候出了什么纰漏……
P.S. 英文维基上查到的5-氨基四唑的密度是1.502 g/cm^3……

| 时段 | 个数 |
|---|---|
| {{f.startingTime}}点 - {{f.endTime}}点 | {{f.fileCount}} |


200字以内,仅用于支线交流,主线讨论请采用回复功能。